

Hemos preparado este artículo para profesionales de la salud que se puedan encontrar ante la posibilidad de un diagnóstico del Síndrome de Rokitansky (medicos/as de familia, pediatras, ginecólogos/as, enfermeras). Al tratarse de un síndrome poco habitual, el mal diagnóstico ocurre con frecuencia, y determina la manera en que la paciente, se aceptará como mujer. Por favor, profesional de la salud, lee, comparte, pregunta, deriva, sé amable!
Te resumimos con un video, de qué se trata el síndrome y lo importante que eres en nuestro diagnóstico. No somos médicos, sino mujeres con el síndrome. Y te lo contamos desde nuestras experiencias.
A continuación del video, encontrarás detalles más técnicos sobre el Síndrome de Rokitansky. También puedes consultar nuestra sección de FAQs (preguntas frecuentes)
Créditos del vídeo:
- Guion y edición vídeo: Paula Rey @estoquesoy.es
- Edición sonido: Gius Gius www.taakstudio.com
1. ¿QUÉ ES?
El síndrome Mayer Rokitansky Küster Hauser (MRKH) es un grupo de anomalías congénitas graves, en raras ocasiones adquiridas, del tracto reproductor femenino, con ausencia de menarquia e imposibilidad de embarazo.
Estas anomalías consisten en la ausencia del útero, trompas de Falopio y del tercio superior de la vagina.
2. DESARROLLO
2.1. ORIGEN Y TIPOS
El aparato reproductor femenino consta de vagina, útero, trompas de Falopio y ovarios. Ilustración 1.
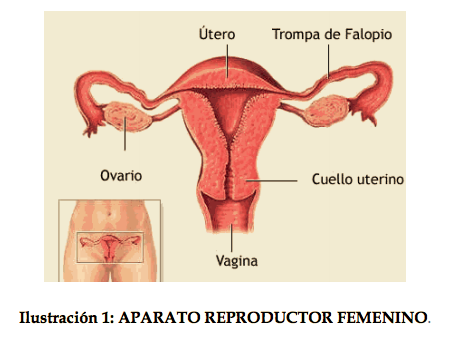
Su origen viene dado por un defecto del desarrollo, fusión o canalización de los conductos de Müller en la etapa embrionaria, excepcionalmente pueden deberse a una alteración adquirida. Los conductos de Müller son dos conductos embrionarios que darán lugar a la formación del útero, vagina y trompas de Falopio en las mujeres y que en el caso de los hombres se atrofian quedando únicamente los conductos de Wolff. En la ilustración 2 se pueden distinguir los diferentes conductos

Las pacientes de dicho síndrome poseen ovarios normales tanto en estructura como en función, ovulando cada mes una vez llegado el periodo fértil. Y, aunque el funcionamiento de los ovarios es totalmente normal y sin ninguna variación respecto a los de una mujer sin el síndrome, no se produce el sangrado propio de la menstruación, ya que al no tener útero ni tercio superior de la vagina, no se forma el endometrio que es el que da lugar al sangrado común cada 28 días.
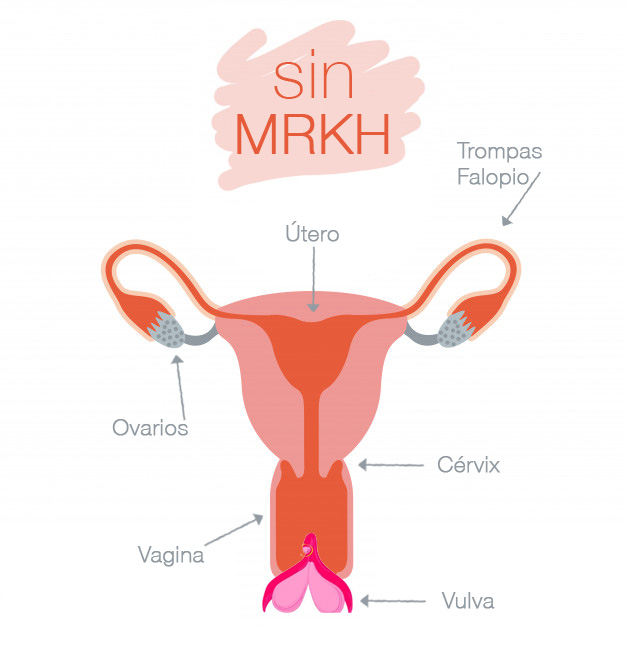
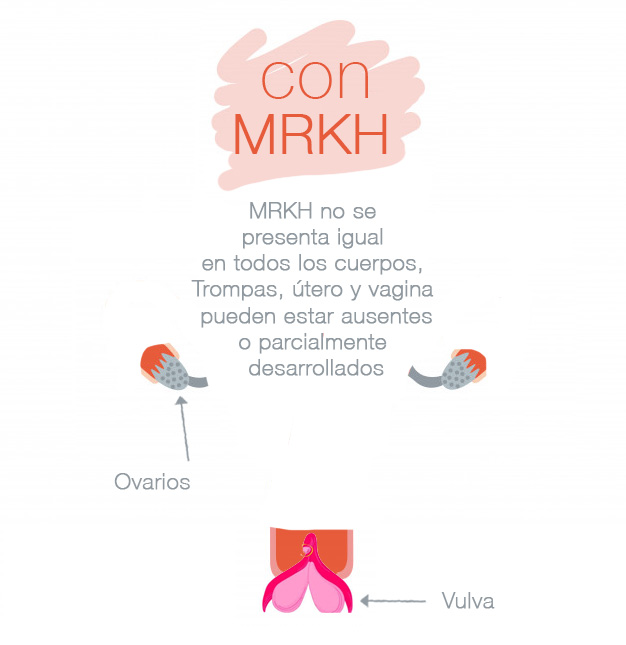
Existen dos tipos:
– MRKH tipo I en el que se da una aplasia completa. Aplasia del útero y tercio superior de la vagina con presencia totalmente normal de trompas de Falopio y ovarios.
– MRKH tipo II en el que se pueden diferenciar malformaciones que afectan no solo al aparato reproductor sino a otros aparatos o sistemas cercanos, como son ciertas malformaciones en el aparato renal superior, malformaciones esqueléticas, problemas de audición y algunas malformaciones cardiacas, siendo estas últimas las más inusuales.
2.2. EPIDEMIOLOGÍA
El rango de incidencia es de 1 mujer entre 4.500 recién nacidos del sexo femenino, sobre un 0,022% aproximadamente. La gran mayoría de los casos diagnosticados son casos aleatorios, es decir, no están relacionados con ningún gen, aunque sí es cierto que se han encontrado casos reiterados en una misma familia.
2.3. PRINCIPALES TÉCNICAS DE DIAGNÓSTICO
La principal razón del diagnóstico es la amenorrea. Cuando con 15 o 16 años una mujer no ha experimentado la menarquia, acude a un especialista y es ahí mediante una exploración vaginal o bien por imagen (ecografía) se observa la ausencia del tercio superior de la vagina y del útero.
Estas diferencias se pueden observar única y exclusivamente en el aparato reproductor interno, los genitales externos tienen apariencia totalmente normal, al igual que los caracteres sexuales secundarios tales como, mamas, vello púbico, etc. Esto se debe a que el sistema hormonal sexual de la mujer funciona con normalidad, por lo que las hormonas responsables del desarrollo de los caracteres secundarios están presentes en la mujer.
Es por esto que hasta la adolescencia no se suele detectar, si no existe otro problema asociado que salga antes a la luz,.
La técnica de diagnóstico por imagen que más se usa es la ecografía pélvica, ya que esta se puede realizar fácilmente en la consulta ginecológica. Esta prueba puede ayudar a reconocer estructuras similares al útero, identificar ovarios, lo cual es fundamental en el descarte de algunos diagnósticos diferenciales, y evaluar otros órganos, como los riñones, dado que este síndrome puede asociarse a malformaciones del sistema urinario. El motivo por el que otros órganos, como los riñones o el sistema renal en general, pueden estar afectados por este síndrome es debido a que estas se forman en el mismo momento en la formación del feto.
El método de diagnóstico que recomendamos es una resonancia magnética (RM) o la Tomografía Axial Computarizada (TAC), que es un examen no invasivo, en el cual no se emplea radiación ionizante, con una alta resolución de contraste, y el más recomendable, que permite la evaluación multiplanar de las malformaciones müllerianas.
El médico responsable de esta prueba diagnóstica, debe estar familiarizado con la representación de las malformaciones anatómicas del aparato reproductor femenino en la resonancia magnética para poder realizar un buen diagnóstico diferencial, y por lo tanto, un adecuado tratamiento.
Para el correcto diagnóstico de este síndrome, y para evitar confusiones con otras enfermedades, se acompañan las técnicas diagnósticas que se van a nombrar a continuación con este cuadro, que por descarte, ayuda al médico responsable a diagnosticar. Tabla 1.
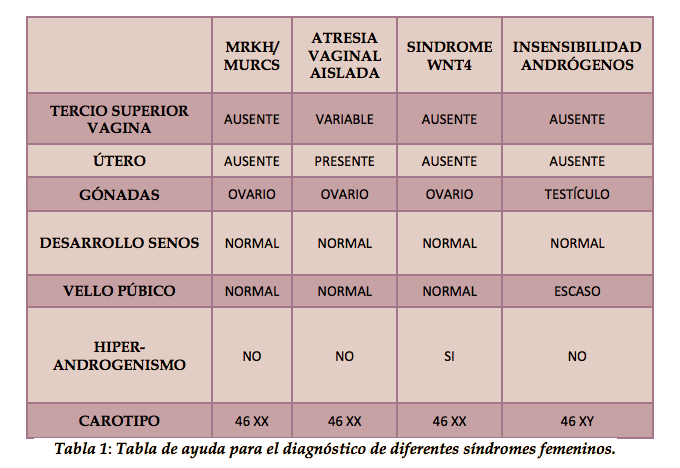
Una prueba genética, ayudaría a descartar la insensibilidad a los andrógenos. El hiperandrogenismo nos hará decantarnos o no por el síndrome WNT4. Ya solo nos quedaría el MRKH y la atresia vaginal aislada, las cuales distinguiremos por la presencia o ausencia de útero.
Es importante destacar que este cuadro es simplemente para facilitar el diagnóstico, pero que deberá ir acompañado de todas las pruebas diagnósticas por imagen, además de otras complementarias como análisis genéticos.2.4. MALFORMACIONES ASOCIADAS
2.4.1. Malformaciones del tracto urinario superior Este tipo de malformaciones se encuentran en, aproximadamente, el 40% de los casos de MRKH. La característica principal es el mal desarrollo del aparato renal (24-28%) , así como la ectopia (mala ubicación) de uno o ambos riñones (17%), hipoplasia renal (desarrollo incompleto) ( 4%) y riñón en herradura en muchos casos.
2.4.2. Anomalías esqueléticas Estas anomalías están principalmente relacionadas con problemas de la columna vertebral (30-40%). Algunas anomalías asociadas son la escoliosis (desviación de la columna vertebral); o anomalías vertebrales aisladas como vertebras asimétricas, condensadas o vertebras en cuña.
Otra anomalía que podemos asociar a este síndrome es la fusión de al menos dos segmentos cervicales, lo que no permite un correcto movimiento del cuello limitando los movimientos. Se ha hallado relación también con casos de malformaciones costales o espina bífida.
2.4.3. Deficiencia auditiva Los defectos de audición o sordera se encuentran en un 10-25% de los pacientes con MRKH. Estos defectos se refieren a sordera conductiva por malformaciones dadas en el oído medio. Algunas de estas enfermedades situadas en el odio medio son: la anquilosis estapediovestibular, que se da por la limitación de movimiento en las articulaciones del conducto; y los defectos neurosensoriales de gravedad variable.
2.4.4. Malformaciones cardiacas No es muy común asociar malformaciones cardiacas al síndrome MRKH, aunque las que se han relacionado han necesitado cirugía lo antes posible. Algunas malformaciones fueron: ventana aorto-pulmonar, fibrilación defecto septal, defectos conotruncales o tetralogía de Fallot. La más frecuente es la ventana aorto-pulmonar la cual consiste en la comunicación que se produce entre, como su nombre indica, la arteria pulmonar y la arteria aorta ascendente.
2.5. ETIOLOGÍA
El síndrome Mayer Rokitansky no se relaciona con ningún factor genético, si bien es cierto que, se han encontrado varios casos en una misma línea familiar. Al no estar éste relacionado con ningún aspecto genético, se valoran las hipótesis de que esté relacionado con factores ambientales o bien con tratamientos médicos.
Algunas de estas causas pueden ser la diabetes gestacional o el uso de talidomida por parte de la madre, la cual se ha demostrado que es teratógena.
Al revisar historiales de embarazos cuyos nacimientos tenían este síndrome, se pudo concluir que no existía relación entre el consumo de medicamentos o drogas por parte de la madre o su exposición a agentes teratógenos con el desarrollo de este síndrome.
La segunda hipótesis que se barajó es la posibilidad de una transmisión por herencia poligénica o multifactorial, es decir, que este síndrome no solo viene determinado por un gen, sino por la unión de varios de ellos.
Otra de las hipótesis que se barajan, es la mutación del gen de la hormona antimülleriana (AMH), o el gen del receptor de la misma.
Esta hipótesis es la más seguida actualmente y hacia la que apuntan todos los estudios, ya que una mutación en dicho gen, afectando a la ruta de la AMH, se puede dar tanto por un gen de transmisión autosómica dominante como se ha explicado anteriormente, como por los diruptores hormonales como se verá en apartados posteriores.
Esto puede provocar que esta hormona se produzca fuera de tiempo, cuando el receptor pueda responder a la hormona y no cuando debería producirse. Pero, ¿por qué es tan importante la AMH? Pues bien, durante la gestación influyen dos tipos de hormonas en la diferenciación sexual del feto.
La primera hormona influyente sería la testosterona, que es la responsable del desarrollo de los conductos deferentes, vesículas seminales, próstata, epidídimos, genitales externos masculinos, etc. Esta hormona no se encuentra alterada en las pacientes de MRKH, ya que sus genitales y caracteres sexuales secundarios se encuentran bien desarrollados acorde a su sexualidad.
La segunda hormona influyente en el desarrollo es la AMH. La AMH es una hormona producida por las células de Leydig y Sertoli, que son las células somáticas testiculares. Esta hormona posee un papel muy importante como es producir la regresión de los conductos de Müller; de ahí que adquiera este nombre. Los conductos de Müller son el inicio embrionario del útero y trompas de Falopio.
3. TRATAMIENTO
– ANATÓMICO Respecto al tratamiento que se ofrece a las personas que padecen este síndrome, podemos dividirlo en dos grandes grupos: tratamiento no quirúrgico y tratamiento quirúrgico.
La elección de un método u otro se realiza de manera conjunta entre médico y paciente, teniendo en cuenta las ventajas que poseen un tratamiento respecto al otro en el caso particular de esa paciente.
DILATACIÓN – En las pacientes en las que existe 1/3 del resto vaginal externo, pero no el 2/3 superior ni el útero, lo primero que habría que hacer es valorar dilatarlo, ya que es el mejor tejido. Ya está en su sitio, tiene mucosa vaginal, hidratada, no hay cicatrices en la entrada… Y el tejido humano permite estiramientos muy importantes, si es poco a poco. La limitación es que es lento, y se ha de ser constante. Pero no provoca dolor, y hoy en día entre los dilatadores metálicos y los de silicona, se consigue mucho. CIRUGIA – Si hablamos de intervención quirúrgica, una manera un poco más rápida y menos dependiente de la voluntad y constancia de la mujer es la dilatación según la técnica de Vecchietti. En este caso es una intervención por laparoscopia. Las ténicas más utilizadas son las de Vecchietti, Wharton o Davidov. Dependerá de la anatomía de la paciente y si ya ha sido operada previamente.
En las pacientes en las que realmente no hay ningún tipo de resto de tercio de la vagina, hay que hacer la neovagina con otras técnicas. creando espacio y con injertos libres o de piel sinténtica, o una vagina con colon.
– EMOCIONAL Abordar la parte emocional es tan o más prioritaria que la anatómica. Principalmente en el momento del diagnóstico, ya que seguramente estaremos ante una adolescente. Recomienda a mamás y papás asistencia emocional. Recomienda que contacten con AMAR. Ya que tenemos un grupo de Whatsapp para mamás y papás y otro grupo para mujeres con el síndrome. Es crucial compartir experiencias y recibir apoyo de mujeres que hayan pasado por el tratamiento o estén en ello.
Y como profesional de la salud ten en cuenta que:
- Soy persona. Soy mujer. No un cliente ni conejillo de indias.
- Si no sabes mucho sobre el síndrome, no simules hacerlo con prepotencia y soberbia. Es normal, no podemos saber todos de todo. Dime que vuelva otro día y consúltalo e investiga.
- DIME QUE PODRÉ SER MADRE , QUE PODRE VIVIR MI SEXUALIDAD PLENAMENTE Y QUE NADIE TIENE QUE SABER QUE TENGO EL SÍNDROME SI NO LO DIGO.
- Háblame con claridad y dime la verdad. Si tu la omites, yo viviré una mentira.
- No sientas pena. Yo soy mujer, aunque sentada fente a tí me vea como una niña. Haz que sienta que me crees y ves como una mujer. Es muy importante.
- Cuídame. Si estás sentado/a frente a mí, es porque tú lo has elegido.


